
La trasformazione batterica: cattura di un gene per diventare “fluo”
-
Biologia
-
Classi: 5° anno
-
-
-
Laboratorio attrezzato
-
Esperimento
-
4 h
-
Min. 2 persone
-
Richiede precauzioni
Riassunto / Abstract
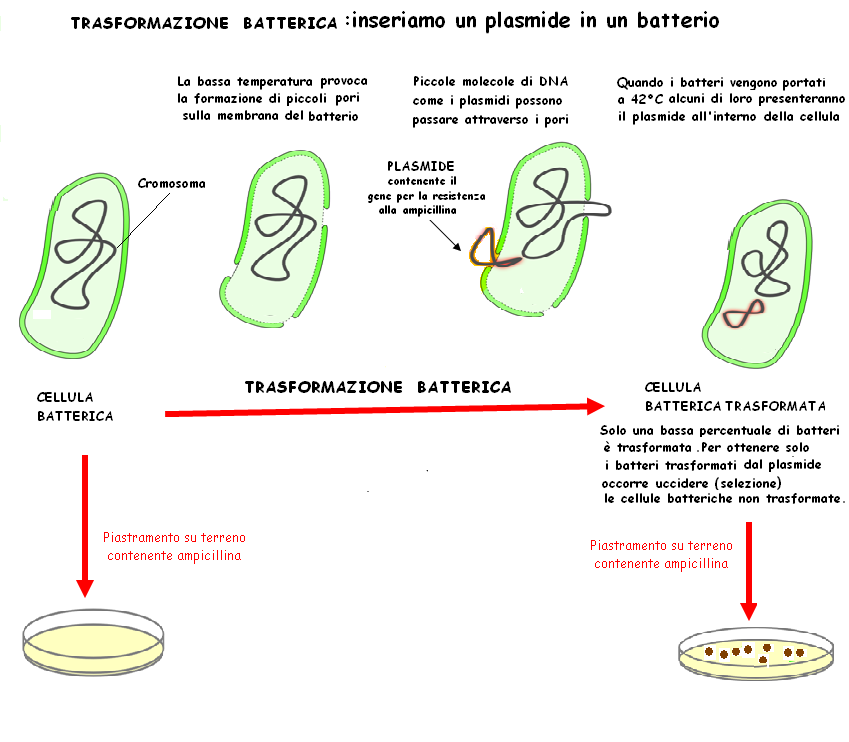
Gli studenti eseguono una procedura, nota come trasformazione batterica, che consiste nell’acquisizione di materiale genetico dall’ambiente esterno. In seguito al processo di trasformazione con un plasmide contenente il gene che codifica per la proteina responsabile del fenomeno della fluorescenza (GFP green fluorescens protein ) della medusa Aequorea victoria, i batteri emettono una luce verde brillante
Scheda sintetica delle attività
- Preparazione di piastre starter ( LBA ) di E. coli ricevente (DH10b) da rendere competente per la trasformazione ed incubare la a 37°C per 18-24 ore. Questa fase è a cura del docente.
- Preparazione di cellule competenti : risospendere 1- 2 colonie di DH10b in 0,25 ml di soluzione trasformante (freddo)
- Trasformazione:aggiungere il DNA plasmidico alla provetta contenente le cellule competenti. Mettere la provetta contenente DNA +cellule competenti in ghiaccio per 10 minuti quindi trasferire la provetta a 42° C per 50 secondi ( shock termico). Porre di nuovo la provetta in ghiaccio per 2 minuti. Aggiungere 0,25 ml di LB sterile ed incubare a temperatura ambiente per 10 minuti.
- Piastramento trasferire una aliquota (0.1 ml) in piastre contenenti antibiotico ( ampicillina) per selezionare i batteri resistenti che hanno ricevuto il plasmide. Incubare per 24h a 37°C.
Risorse necessarie
- Escherichia coli DH10b per fare cellule competenti*;
- plasmide pJBA27 60 nanogrammi /microlitro*;
- ampicillina (50 mg /ml );
- soluzione di trasformazione (50 mM CaCl2,), sterile, 5 ml;
- brodo nutriente LB, sterile, 10 ml;
- brodo nutriente LB+agar.sterile 500ml;
- pipette sterili;
- anse per inoculare, sterili, da 10 μl;
- capsule Petri, 60 mm;
- microprovette, sterili, da 1.5 ml e relativo porta provette;
- spatole sterili monouso o spatole di vetro;
- pennarelli indelebili;
- lampada U.V.—ampia lunghezza d’onda (non indispensabile);
- cronometro od orologio o cellulare;
- bagno termostatato o pentolino con acqua;
- termometro per misurare 42°C;
- ghiaccio tritato e contenitori (p.es., bicchieri di plastica ).
* Lo specifico ceppo di E. coli e il plasmide contenente il gene che codifica per la GFP possono essere forniti tramite posta su richiesta degli interessati.
Prerequisiti necessari
Conoscenze teoriche:
- struttura e funzioni della cellula procariota;
- crescita batterica;
- controllo della crescita batterica: metodi di sterilizzazione con agenti fisici o chimici;
- struttura e funzioni del DNA;
- plasmidi.
Conoscenze-pratiche:
- preparazione di terreni di coltura;
- preparazione di soluzioni;
- capacità di operare in condizioni di asepsi per evitare contaminazioni con batteri presenti nell'ambiente di lavoro.
Obiettivi di apprendimento
- Esprimere i concetti acquisiti con lessico appropriato;
- acquisire consapevolezza dei principi di base della microbiologia e della necessità mantenere le colture pure per effettuare esperimenti mirati ad analizzare le caratteristiche fenotipiche / genetiche di un determinato ceppo batterico;
- individuare gli elementi e gli aspetti rilevanti del fenomeno;
- saper organizzare l'attività di laboratorio;
- saper interpretare e commentare i dati sperimentali.
Dotazioni di sicurezza
Il ceppo DH10b è un Escherichia coli K–12 non patogeno, il plasmide contenente il gene della GFP ricombinante e le cellule trasformate originatesi dalla loro combinazione non sono organismi patogeni. Tuttavia, maneggiare le cellule derivanti dalla trasformazione, implica l’applicazione di regole standard di microbiologia.
Svolgimento
Fase preparatoria
- Terreno di cultura: fare riferimento all'esperimento 102-Scienze.
- Enterogermina: miliardi di spore di Bacillus clausii da bere. Allestire una serie di piastre contenenti 50µg/ml di ampicillina aggiungendo X ml di una soluzione madre di Ampicillina 5mg/ml l utilizzando la seguente formula: $$C_iV_i = C_fV_f$$
Dove:
$C_i$ è la concentrazione iniziale (soluzione madre 5mg/ml ovvero5000µg/ml);
$V_i$ è l’incognita della formula;
$C_f$ è la concentrazione finale di Ap (50µg/ml);
$V_f$ è il volume finale (es 400ml di LBA per circa 20 piastre).
$V_i$ è l’incognita della formula;
$C_f$ è la concentrazione finale di Ap (50µg/ml);
$V_f$ è il volume finale (es 400ml di LBA per circa 20 piastre).
Quindi, se si preparano 400 ml di terreno: $$V_i = 50 \cdot \frac{400}{500}= 4\ ml$$
- Preparare una soluzione sterile di CaCl2 50 mM.
- Preparare LB sterile.
TRASFORMAZIONE BATTERICA (gruppi di 2 persone)
La trasformazione batterica con plasmidi è una tecnica di biologia molecolare, largamente utilizzata nei laboratori, per facilitare l'introduzione di plasmidi nei batteri. La trasformazione si ottiene modificando alcune proprietà chimico fisiche delle pareti e delle membrane cellulari con l'impiego di sostanze chimiche (CaCl2), associate a rapidi sbalzi di temperatura ne deriva così una temporanea permeabilizzazione delle cellule al DNA esogeno. I batteri contenenti il plasmide esogeno vengono quindi fatti crescere su terreni selettivi con conseguente formazione di colonie trasformate.
La procedura di trasformazione utilizzata dagli studenti si basa essenzialmente su un protocollo pubblicato nel 1972 da Cohen et al. tuttora utilizzato nei laboratori di ricerca. Tale protocollo è stato semplificato evitando l'uso di apparecchiature ( microcentrifuga ) e accorciando i tempi di esecuzione. Tuttavia i risultati sono quelli ottenibili nei laboratori di ricerca anche se l 'efficienza di trasformazione (numero di trasformanti ) è minore
FASE1 Preparazione “piastra starter”
(da eseguire il giorno precedente all'esperimento di trasformazione)
La piastra starter deve essere preparata e incubata a 37°C 24-36 ore prima dell’inizio dell’esperimento di trasformazione e non va messa in frigo. Anche nei laboratori di ricerca, nei quali la procedura è leggermente più lunga e prevede l’uso di centrifughe, per eseguire un esperimento di trasformazione batterica si utilizzano colture “fresche” ovvero in una particolare fase di crescita ( fase di crescita esponenziale ).
La piastra starter deve essere preparata e incubata a 37°C 24-36 ore prima dell’inizio dell’esperimento di trasformazione e non va messa in frigo. Anche nei laboratori di ricerca, nei quali la procedura è leggermente più lunga e prevede l’uso di centrifughe, per eseguire un esperimento di trasformazione batterica si utilizzano colture “fresche” ovvero in una particolare fase di crescita ( fase di crescita esponenziale ).
Mentre Le piastre per la trasformazione contengono l’ampicillina 50 microgrammi/ml in quanto il plasmide utilizzato possiede sia il gene per la GFP che il gene per la resistenza alla ampicillina Le piastre starter per la preparazione delle cellule di E. coli da trasformare non contengono antibiotico Poiché il ceppo DH10B è resistente alla streptomicina ( questa resistenza non è plasmidica ma cromosomale per evitare problemi di contaminazione si possono preparare piastre starter contenenti streptomicina 100 microgrammi/ml (LBA Sm100)
La piastra starter di E. coli DH10b il ceppo ricevente sensibile all’ ampicillina in quanto privo del plasmide pJBA27 deve essere preparate in modo da ottenere singole colonie. Questa operazione va eseguita con molta attenzione per evitare contaminazioni: si preleva con una ansa sterile il ceppo DH10b (da una brodocultura o piastra “vecchia” che può essere conservata per settimane a 4°C) e si distribuisce uniformemente sulla superficie dell’agar (LBA o LBA Sm100). Inserire un’ansa sterile per inoculare direttamente nella sospensione batterica. Estrarre l’ansa e inoculare le piastre come illustrato. L’inoculo si esegue sequenzialmente, dividendo la piastra in 4 quadranti e va effettuato strisciando l’ansa avanti e indietro per una dozzina di volte sull’area di ciascun quadrante. La prima strisciata (A) serve per spargere un poco le cellule. Ripetendo l’operazione nei quadranti successivi, i batteri vengono via via sempre più diluiti, aumentando la probabilità di ottenere singole colonie. Per le strisciate si deve usare il massimo della superficie dei quadranti. Ruotare la piastra di ~45° (così da poter muovere comodamente l’ansa) e procedere con la seconda strisciata. Passare sulla strisciata precedente un paio di volte e poi altre 10 sulla superficie del secondo quadrante (B) Ripetere il punto b sul terzo quadrante(C) e sul quarto quadrante( D)
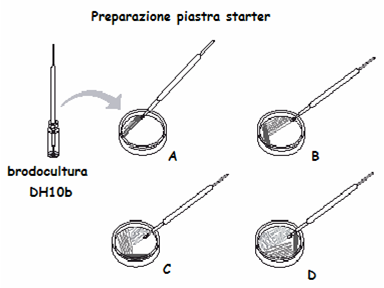
Mettere le piastre "starter" capovolte nell’incubatore a 37°C per tutta la notte o a temperatura ambiente per 2–3 giorni, qualora l’incubatore non fosse disponibile. Usare la piastra starter per la trasformazione entro 24–36 ore NON METTERE IN FRIGORIFERO
FASE 2: Trasformazione
Ogni gruppo di studenti necessita di una piastra starter (coltura ricevente) da cui prelevare le cellule da trasformare.
La procedura di trasformazione si svolge principalmente in tre momenti, che hanno lo scopo di introdurre il plasmide nelle cellule di E. coli e di creare un ambiente nel quale i batteri possano esprimere il gene acquisito.
- Preparazione di cellule competenti : Escherichia colinon è naturalmente competente per la trasformazione. Tuttavia tramite trattamento con cloruro di calcio e shock termico ed utilizzando batteri in una specifica fase di crescita è possibile rendere il batterio permeabile all'ingresso di DNA plasmidico di piccole dimensioni. Risospendere 1- 2 colonie di DH10b in una provetta da 1.5 ml contenente 0,25 ml di soluzione trasformante ( 50 mM CaCl2 freddo). Ogni gruppo deve preparare 2 aliquote di cellule competenti: una contrassegnata con la scritta +Pla (alla quale si aggiungerà il DNA plasmidico) ed una -Pla per il controllo negativo senza DNA.
- Trasformazione mediante shock termico :aggiungere il DNA plasmidico(100-200 nanogrammi) solo alla provetta contenente le cellule competenti contrassegnata con la scritta +Pla. Mettere entrambe le provette in ghiaccio per 10 minuti quindi trasferire le provetta a 42° C per 50 secondi (shock termico). Porre di nuovo la provetta in ghiaccio per 2 minuti.
- Rivitalizzare i batteri Occorre ripristinare le funzioni metaboliche fornendo nutrienti per permettere la produzione della proteina per l’ampicillina–resistenza A tale scopo aggiungere 0,25 ml di terreno nutritivo sterile( LB) ed incubare a temperatura ambiente per 10 minuti. .
Trascorsi i 10 minuti eseguire il piastramento ( figura) ovvero trasferire una aliquota (0.1 ml) in piastre contenenti ampicillina (LBA Ap50 ) per selezionare i batteri resistenti che hanno ricevuto il plasmide ed incubare per 24h a 37°C.
FASE 3 analisi dei risultati
La lettura dei risultati consiste nell'osservare e contare le colonie cresciute sulle piastre LBA Ap50 della trasformazione e sulle piastre LBA Ap 50 di controllo.
Sulle piastre di controllo risultato atteso è ASSENZA di colonie
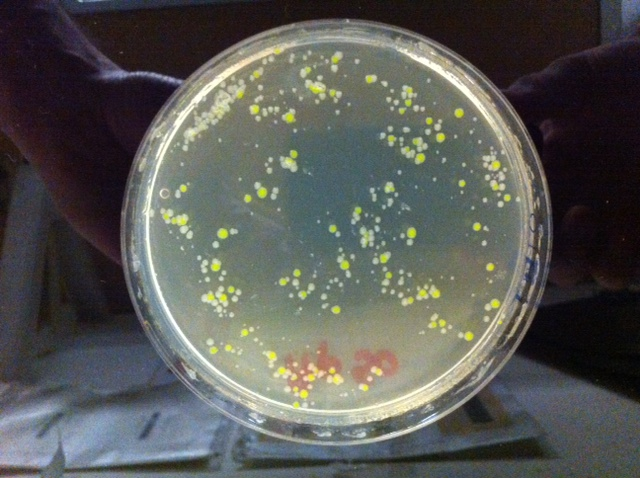
Sulle piastre della trasformazione, in cui è stato aggiunto il DNA plasmidico, si osserveranno numerose colonie di colore verde "abbastanza Fluo" che osservate alla luce ultravioletta risulteranno ancora più Fluorescenti. Spesso intorno alla colonia fluorescente sono presenti piccole colonie non fluorescenti e più piccole (definite "satelliti") che non hanno acquisito il plasmide ma riescono a crescere in presenza di ampicillina grazie all'enzima che distrugge l'ampicillina, prodotto dalle cellule trasformate, il quale diffonde nel terreno. Il numero di colonie trasformate può variare in base al ceppo di E. coli utilizzato ( DH10b è un ottimo recipiente per la trasformazione) e al tipo di plasmide utilizzato. Nel caso specifico si dovrebbero ottenere alcune centinaia di colonie resistenti all'ampicillina e quindi fluorescenti. Il numero di colonie ottenuto dipende dalla più o meno corretta esecuzione dell'esperimento, dalle dimensioni del plasmide e dalla giusta quantità di molecole di plasmide aggiunto. Con il plasmide pJBA27,un plasmide piccole dimensioni (meno di 4000 coppie di basi ) sono sufficienti circa 150 nanogrammi di DNA e quantità maggiori non aumentano l'efficienza di trasformazione. In condizioni ottimali, come quelle descritte, il piastramento diretto di 0.1ml di miscela di trasformazione ha portato alla crescita di numerose colonie sia di trasformanti che di colonie satelliti per cui era difficile effettuare una conta come pure distinguere i veri trasformanti. E consigliabile quindi far piastrare ad alcuni gruppi di studenti sia 0.1 ml della miscela (piastramento diretto) che 0.1 di una diluizione 10-1 Nella figura 3 si può osservare il risultato ottenuto con il piastramento di 0.1 ml della diluizione 10-1 in cui sono ben visibili le colonie trasformanti resistenti alla ampicillina: sono più grandi e poiché il plasmide porta il gene che codifica per la GFP appaiono anche di colore verde fluo. Da notare che la foto è stata scattata senza luce ultravioletta.
Decontaminazione ed eliminazione dei rifiuti
Se non si dispone di un’autoclave, sarà cura dotare ogni banco di lavoro di un apposito recipiente contenente una soluzione di sterilizzazione (10 % di candeggina, preparata al momento). Tutte le soluzioni ed il materiale (anse e pipette) venute in contatto con i batteri devono essere raccolte nel recipiente e lasciate per almeno 20 minuti, al fine di sterilizzarli.
Bisogna sempre sterilizzare le anse e le pipette utilizzate per l’esperimento.
Le capsule Petri vanno sterilizzate versandoci dentro la soluzione di sterilizzazione. Questa, dopo aver agito per almeno un’ora, può essere eliminata direttamente nello scarico. Una volta trattate, le piastre possono essere eliminate come normali rifiuti.
E’ consigliato l’uso di occhiali quando si utilizza la soluzione sterilizzante.
Bisogna sempre sterilizzare le anse e le pipette utilizzate per l’esperimento.
Le capsule Petri vanno sterilizzate versandoci dentro la soluzione di sterilizzazione. Questa, dopo aver agito per almeno un’ora, può essere eliminata direttamente nello scarico. Una volta trattate, le piastre possono essere eliminate come normali rifiuti.
E’ consigliato l’uso di occhiali quando si utilizza la soluzione sterilizzante.
Note e storia
La trasformazione e la scoperta del DNA come materiale genetico
Le infezioni da pneumococco (Streptococcus pneumoniae) rappresentavano prima della scoperta degli antibiotici una delle principali cause di morte. Questo batterio oltre ad essere l’agente eziologico della maggior parte delle polmoniti è responsabile di altre patologie infettive quali sinusiti, congiuntiviti, otiti e forme gravi di meningiti. Nel 1917 Oswald Avery iniziò uno studio sulla biologia dello pneumococco e scoprì che alcune colture di pneumococco liberavano sostanze solubili specifiche (SSS) presenti nelle urine di pazienti affetti da polmonite. Successivamente scoprì che le SSS derivavano da un rivestimento di Streptococcus pneumoniae (la capsula) e trovò una correlazione tra presenza della capsula e virulenza degli pneumococchi: batteri capsulati sono virulenti batteri privi di capsula sono non virulenti.
Questi studi portarono Frederick Griffith a scoprire, nel 1925, che gli pneumococchi isolati da pazienti infetti erano di due tipi: alcuni innocui che davano origine a colonie di aspetto rugoso (fenotipo R da rough ) mentre altri davano origine a colonie di aspetto liscio( fenotipo S da smooth) . Solo le colture derivanti da colonie di tipo S erano virulente ovvero se iniettate nel topo ne causavano la morte. Tre anni dopo Griffith fece una straordinaria scoperta per le conoscenze dell’epoca: l’iniezione di batteri S uccisi al calore come quella di batteri di tipo R , non provocava la morte dei topi; se invece batteri di tipo R vivi venivano iniettati insieme a batteri S uccisi al calore l’animale moriva e da esso venivano isolati batteri di tipo S. Quindi Griffith concluse che un principio trasformante presente nelle cellule S uccise poteva TRASFORMARE le cellule R (avirulente) in S (virulente).
Questo esperimento, fondamentale per gli sviluppi successivi della Biologia, all’epoca non ebbe la giusta risonanza in quanto non era ancora nota la natura del materiale genetico. Per molti microbiologi, l’ osservazione di Griffith appariva difficile da spiegare e poco credibile. Oswald Avery, che era uno di questi, fece ripetere nel 1931 l’esperimento ai suoi collaboratori e ottenne gli stessi risultati di Griffith. Undici anni dopo, lo stesso Avery giunse alla conclusione che il principio trasformante era il DNA in quanto era sufficiente il DNA estratto da pneumococco di tipoS per trasformare un ceppo R avirulento in ceppo S virulento. All’epoca la tecnologia per l’estrazione di DNA era solo all’inizio ed occorrevano decine di litri di coltura batterica per ottenere pochi milligrammi di DNA, inoltre non si poteva escludere nella preparazione la presenza di proteine o polisaccaridi contaminanti in grado di trasformare. In seguito nel 1944 Avery e McCarty osservarono che il trattamento del DNA con DNasi ( un enzima che degrada il DNA) distruggeva l'attività trasformante e quindi dimostrarono che il principio trasformante era il DNA. Nonostante l' importanza di questa scoperta, la pubblicazione dei risultati sulla rivista Journal Experimental Medicine non ebbe grande successo per vari motivi:
- siamo nel dopoguerra gli scambi internazionali tra scienziati erano scarsi;
- la rivista era poco letta dai genetisti interessati a capire la natura del materiale genetico;
- la trasformazione sembrava limitato allo pneumococco ( solo più tardi tale fenomeno è stato osservato in molti altri batteri);
- all'epoca si riteneva che le proteine e non il DNA potessero spiegare la variabilità genetica presente negli organismi viventi.
Finalmente nel 1952 Hershey e Chase, con esperimenti effettuati con i batteriofagi dimostrarono definitivamente che il DNA è la molecola depositaria dell'informazione genetica
Nei batteri la trasformazione batterica insieme ad altri meccanismi di scambio di materiale genetico , quali la coniugazione e la trasduzione, costituisce una importante fonte di variabilità genetica che supplisce alla assenza dei meccanismi di riproduzione per via sessuale presenti negli organismi superiori. La trasformazione naturale è un meccanismo presente solo in alcune specie batteriche: Streptococcus pneumoniae ( ovviamente ), Haemophilus influenzae, Neisseria meningitidis etc... ). Escherichia coli, il microrganismo utilizzato come modello in molti studi, non è naturalmente competente.
Nei batteri la trasformazione batterica insieme ad altri meccanismi di scambio di materiale genetico , quali la coniugazione e la trasduzione, costituisce una importante fonte di variabilità genetica che supplisce alla assenza dei meccanismi di riproduzione per via sessuale presenti negli organismi superiori. La trasformazione naturale è un meccanismo presente solo in alcune specie batteriche: Streptococcus pneumoniae ( ovviamente ), Haemophilus influenzae, Neisseria meningitidis etc... ). Escherichia coli, il microrganismo utilizzato come modello in molti studi, non è naturalmente competente.
La trasformazione artificiale con plasmidi come donatori ed Escherichia coli, reso competenti per la trasformazione, come ricevente è una delle più frequenti tecniche utilizzate nei laboratori di ricerca. I batteri, in aggiunta ad un lungo cromosoma circolare , possono contenere, uno o più strutture di DNA circolari, chiamate plasmidi, in grado di replicarsi autonomamente rispetto al cromosoma . Il DNA plasmidico contiene normalmente geni codificanti per caratteri accessori non indispensabili alla sopravvivenza del batterio, che tuttavia possono conderire al batterio un vantaggio selettivo ( es. la resistenza all'antiotico è un vantaggio se nell'ambiente è presente l'antibiotico) . Gli scienziati usano una metodologia chiamata ingegneria genetica per inserire nei plasmidi geni codificanti per nuovi caratteri. I geni possono essere prelevati dal genoma di qualsiasi organismo−uomo, animali, piante, altri microrganismi−e trasferiti nei batteri. P. es., il gene corretto dell’insulina umana può essere inserito nei batteri i quali, nelle opportune condizioni, possono produrre questo ormone. L’insulina così ottenuta può essere usata per la cura del diabete, patologia causata da un difetto genetico che si traduce nella scarsa o nulla funzionalità della proteina prodotta. Il plasmide pJAB27, utilizzato in questo esperimento è stato costruito introducendo il gene, che codifica per la proteina GFP (GFP green fluorescens protein ) della medusa Aequorea victoria in un plasmide che presenta il gene responsabile della resistenza all’ampicillina.
NOTE
Dotazioni di sicurezza
Il ceppo DH10b è un E. coli K–12 non patogeno, il plasmide contenente il gene della GFP ricombinante e le cellule trasformate originatesi dalla loro combinazione non sono organismi patogeni.Tuttavia, maneggiare le cellule derivanti dalla trasformazione, implica l’applicazione di regole standard di microbiologia. Ciò comporta quanto segue.
1- Le superfici di lavoro devono essere decontaminate giornalmente ed ogni qualvolta si verifichi una contaminazione con materiale biologico.
2-Tutti i rifiuti solidi e liquidi contaminati devono essere decontaminati prima dell’eliminazione.
3-Ci si deve lavare le mani dopo aver lavorato con organismi contenenti molecole di DNA ricombinante e, sempre e comunque, prima di uscire dal laboratorio.
4-Quanto sopra deve essere fatto con attenzione, per ridurre al minimo la produzione di aerosol.
5-Sono ammessi solo apparati per il pipettamento meccanico. E’ vietato pipettare con la bocca.
6-In laboratorio non è ammesso mangiare, bere, fumare e utilizzare cosmetici.
7-E’ consigliato l’uso di occhiali e guanti protettivi.
Decontaminazione ed eliminazione dei rifiuti
Se non si dispone di un’autoclave, sarà cura dotare ogni banco di lavoro di un apposito recipiente contenente una soluzione di sterilizzazione (10 % di candeggina, preparata al momento). Tutte le soluzioni ed il materiale (anse e pipette) venute in contatto con i batteri devono essere raccolte nel recipiente e lasciate per almeno 20 minuti, al fine di sterilizzarli. Bisogna sempre sterilizzare le anse e le pipette utilizzate per l’esperimento. Le capsule Petri vanno sterilizzate versandoci dentro la soluzione di sterilizzazione. Questa, dopo aver agito per almeno un’ora, può essere eliminata direttamente nello scarico. Una volta trattate, le piastre possono essere eliminate come normali rifiuti. E’ consigliato l’uso di occhiali quando si utilizza la soluzione sterilizzante.
Bibliografia
- Cohen SN, Chang AC, Hsu L. 1972 Nonchromosomal antibiotic resistance in bacteria:genetic transformation of Escherichia coli by R-factor DNA. Proc Natl Acad Sci U S A. 69:2110-2114.
Autori
Casalino Mariassunta